


过去5年,国产器械上市总数是进口的15倍,召回占比却不到1/4,是质量太好还是另有隐情?
来源:医休器械平台旗下公众号-医休神介说
作者:医休哥
医疗器械作为一种特殊的商品,在疾病预防、诊断、监护、治疗、缓解等方面具有非常重要的作用,其质量关乎患者健康和生命安全。随着我国经济发展和生活水平提高,居民医疗保健意识增强,临床需求激增,促使器械市场呈现快速增长态势,医疗器械需求不断增加。随着医疗器械批准数量不断增长,医疗器械召回事件数量也逐年上升。
我国的医疗器械召回事件数量从2010年的35例增至2018年的573例,美国的医疗器械召回事件数量也在持续增加,从2003年的650例增到2017年的3202例。我国的医疗器械召回事件数量在2018年达到峰值,在2019—2020年呈逐年下降趋势。
医疗器械上市后监管有助于确保产品的安全性和有效性,2021年新修订的《医疗器械监督管理条例》第二十条明确要求医疗器械注册人、备案人应当开展医疗器械不良事件监测、再评价和建立召回等上市后监管制度。医疗器械召回和追溯制度属于上市后监管的重要一环,医疗器械召回可以控制存在潜在缺陷的医疗器械产品风险,消除医疗器械安全隐患。
2017年,原国家食品药品监管总局发布《医疗器械召回管理办法》,医疗器械召回是指医疗器械生产企业按照规定的程序对其已上市销售的某一类别、型号或者批次的存在缺陷的医疗器械产品,采取警示、检查、修理、重新标签、修改并完善说明书、软件更新、替换、收回、销毁等方式进行处理的行为。
我国医疗器械首次上市的数量与趋势
我国医疗器械产业快速发展,产业营业收入从2019年的0.62万亿元增至2024年的1.35万亿元,年均复合增长率达16.8%,已成为全球医疗器械第二大市场,产业集聚度、国际竞争力不断提升。
2019—2024年,我国首次批准上市的Ⅱ类和Ⅲ类医疗器械数量呈明显上升趋势,从2019年的7937个逐增至2024年的17461个,年均复合增长率达17.1%,Ⅱ类和Ⅲ类医疗器械平均每年注册上市12820个和1953个,见表1。
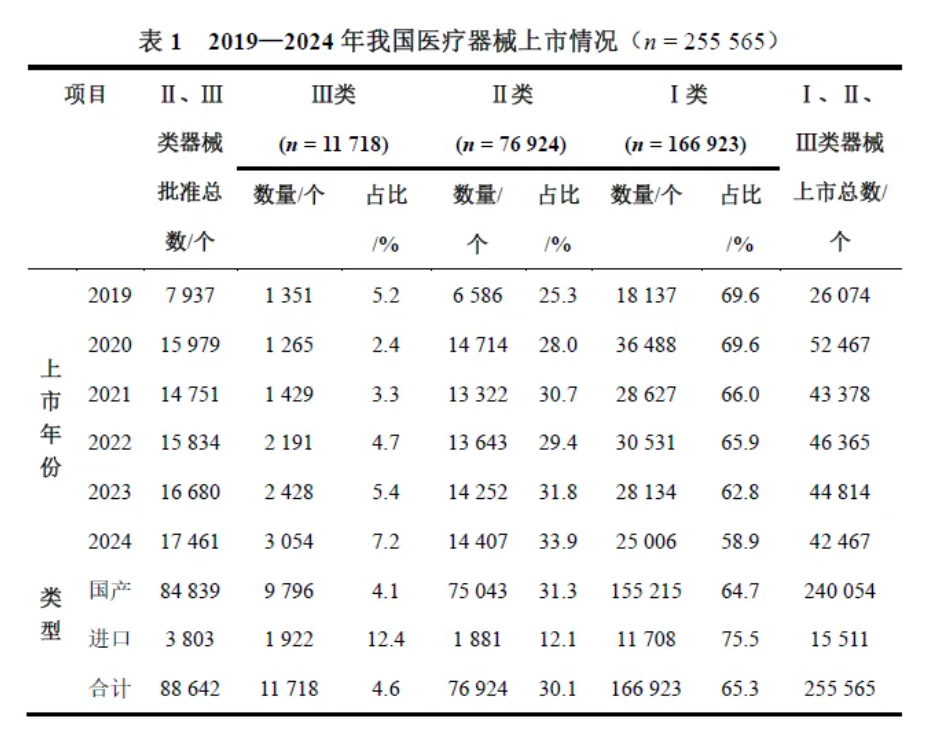
由于受到突发公共卫生事件的影响,2020年后上市了大量的《体外诊断试剂”和《注输、护理和防护器械”的医疗器械,医疗器械备案及注册上市数量增长超过1倍。2020年我国医疗器械上市数量达历史峰值,共52467个,是2019年的2倍。2019—2024年,共上市医疗器械产品255565个,其中Ⅰ类医疗器械占比65.3%,Ⅱ类医疗器械占比30.1%,Ⅲ类医疗器械占比4.6%。
2019—2024年,我国上市的医疗器械总数量是进口的15.5倍。2019—2024年,我国上市的医疗器械中94%为国产医疗器械,进口医疗器械仅占6%。80%的高端医疗设备市场份额被欧美等跨国公司垄断,一些尖端领域甚至完全受制于跨国公司。在进口医疗器械中的Ⅲ类器械比例比国产医疗器械高,国产医疗器械中Ⅲ类器械占全部国产器械上市的4.1%,在进口医疗器械中,Ⅲ类医疗器械占12.4%,见表1。在Ⅲ类医疗器械中,进口医疗器械占16%,见图1。
创新医疗器械上市的数量与趋势
2018年,国家药品监督管理局发布《创新医疗器械特别审查程序》,此后我国创新医疗器械产品获批数量逐年上升。国家药品监督管理局将创新器械定义为具有中国发明专利,技术上属于国内首创、国际领先,具有显著临床应用价值的第Ⅱ和第Ⅲ类医疗器械。国家药品监督管理局医疗器械技术审评中心设立创新医疗器械审查办公室,负责组织专家对创新医疗器械特别审查申请进行审查,对预审查建议、专家意见进行确认,对拟批准的申请进行公示。为加快创新医疗器械上市,国家药品监督管理部门设置特别审批通道,进行优先审评。
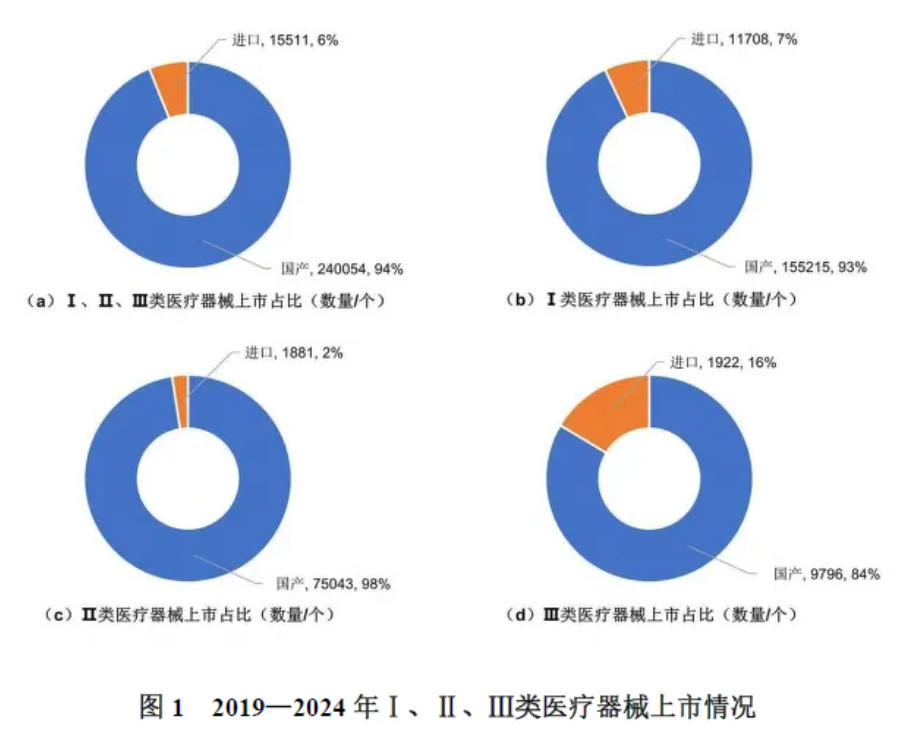
根据2019—2024年度医疗器械注册工作报告显示,创新医疗器械上市数量逐年增加,从2019年的19个增至2024年的65个,年均复合增长率达27.8%。2019—2024年,国家药品监督管理局共收到创新医疗器械特别审批申请1885项,批准上市的创新医疗器械共261项,申请获批率为13.8%。2019—2024年获批的创新医疗器械98.5%为Ⅲ类医疗器械,其中国产医疗器械占比83.9%,产品类别主要集中于无源植入器械、有源手术器械、医用成像器械、有源植入器械和医用软件。
我国医疗器械上市后召回情况
1.医疗器械召回级别及产地特征
根据2017年国家药品监督管理局发布医疗器械召回管理办法》,按照器械的缺陷严重程度,将医疗器械召回分为3个级别:
·一级召回是指使用该医疗器械可能或者已经引起严重健康危害的;
·二级召回是指使用该医疗器械可能或者已经引起暂时的或者可逆的健康危害的;
·三级召回是指使用该医疗器械引起危害的可能性较小但仍需要召回的。
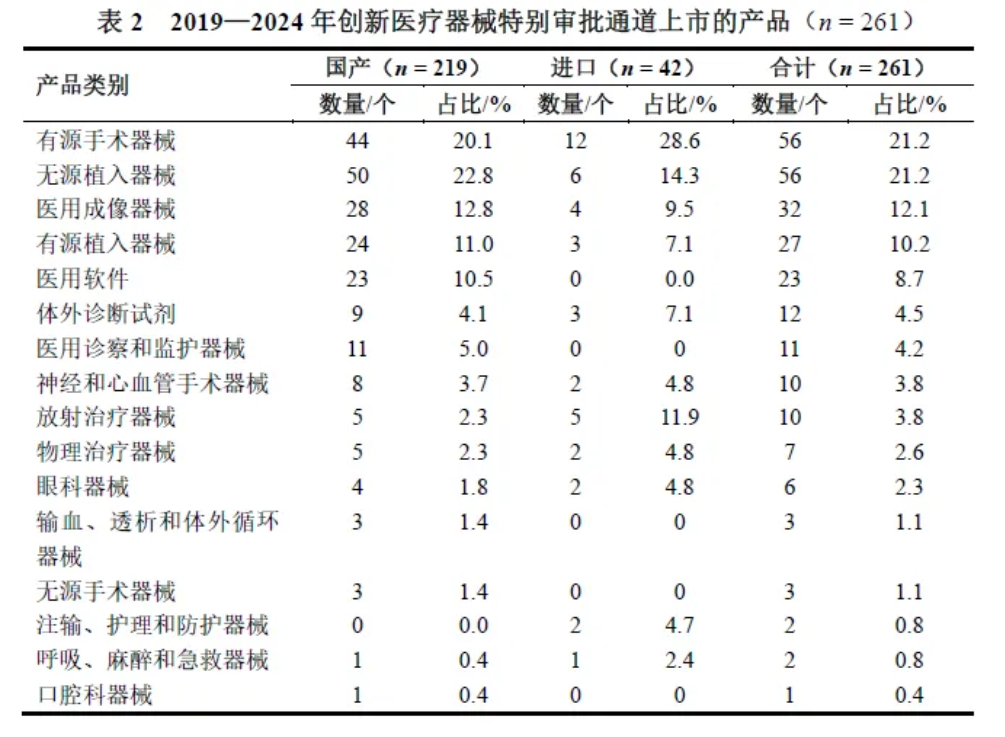
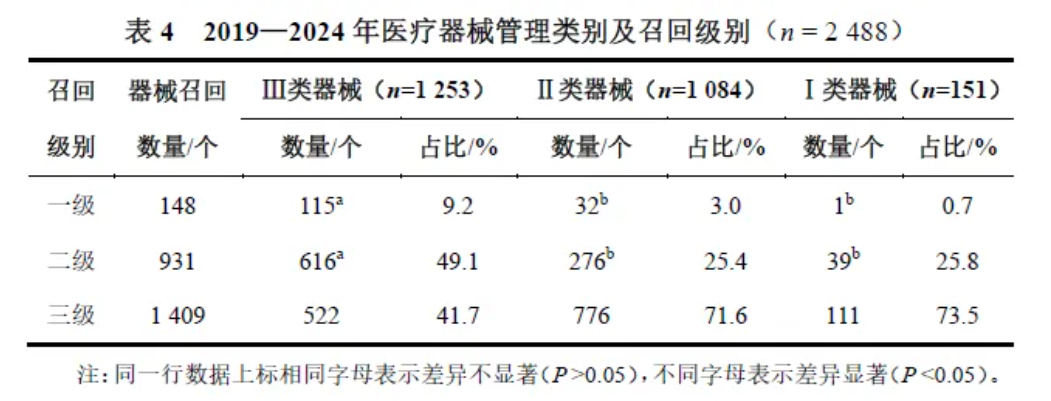
2019年起,我国发生召回的医疗器械数量逐年降低,下降比例达71.5%。2019—2024年发生召回的医疗器械共2488个,每年平均发生召回的医疗器械415个。其中,三级召回医疗器械1409个(56.7%),二级召回931个(37.4%),一级召回148个(5.9%)。
从产地分类来看,进口医疗器械召回占比75.1%,多于国产医疗器械召回。
在国产医疗器械召回中发生最多的是三级召回,占比达94.2%;在进口医疗器械召回中发生最多的是二级召回,占比达48.2%。在一级召回和二级召回中,进口医疗器械比国产医疗器械发生召回的可能性更高(P<0.05),见表3。
2.医疗器械管理类别及召回级别
从管理类别看,在召回的医疗器械中Ⅲ类医疗器械占比最高,其次为Ⅱ类和Ⅰ类医疗器械,分别占比50.4%、43.5%、6.1%。从召回级别看,Ⅲ类医疗器械比Ⅱ类医疗器械以及Ⅰ类医疗器械更有可能发生一级召回和二级召回(P均<0.05)。
3.医疗器械召回的产品类别
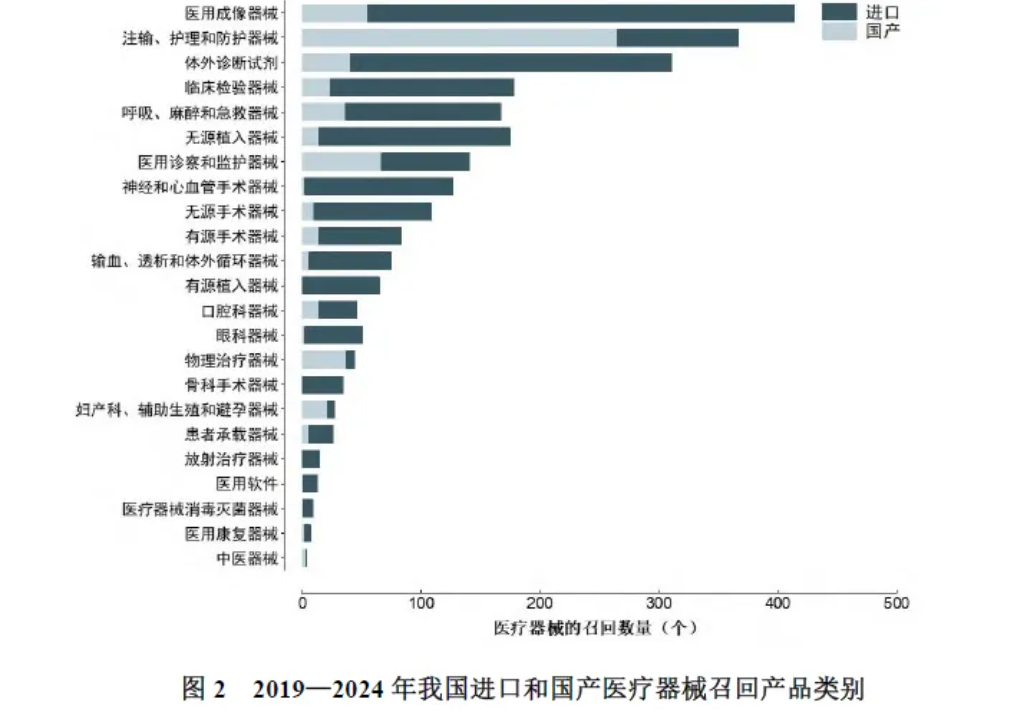
依据2017年原国家食品药品监督管理总局修订的医疗器械分类目录》和2024年国家药品监督管理局修订的《体外诊断试剂分类目录》划分产品类别,医疗器械召回类别最多的是医用成像器械、注输、护理和防护器械以及体外诊断试剂,分别占比16.6%、14.8%、12.5%。国产医疗器械召回最多的是注输、护理和防护器械类产品,进口医疗器械召回最多的是医用成像器械类产品。
医疗器械召回风险发现途径
医疗器械生产企业是控制与消除产品缺陷的责任主体,企业发现存在缺陷的医疗器械产品后经过调查评估发起主动召回程序。当药品监督管理部门认为企业应召回存在缺陷的医疗器械产品而未主动召回的,可以责令其召回。根据医疗器械召回的启动主体不同,医疗器械召回可分为主动召回和责令召回。在实际发生的医疗器械召回中,以企业实施主动召回为主,以药品监督管理部门责令召回为辅。
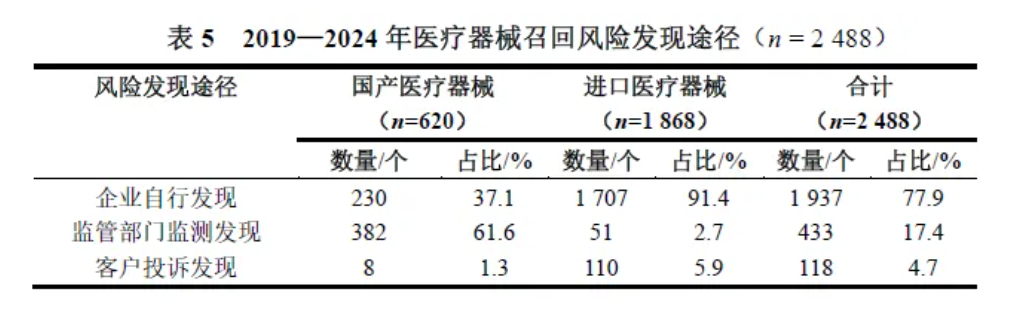
在医疗器械召回风险发现途径方面,2019—2024年医疗器械召回有77.9%为企业自行发现潜在风险后召回,17.4%为监管部门监测发现不合格后召回,4.7%为客户投诉发现问题后召回,见表5。在国产医疗器械召回原因中,61.6%是监管部门监测发现不合格后召回;在进口医疗器械召回原因中,91.5%是企业自行发现潜在风险后召回。
Ⅲ类医疗器械召回特征分析
Ⅲ类医疗器械最易发生召回,在所有召回中占50.4%,在危害程度最高的一级召回中占77.7%。
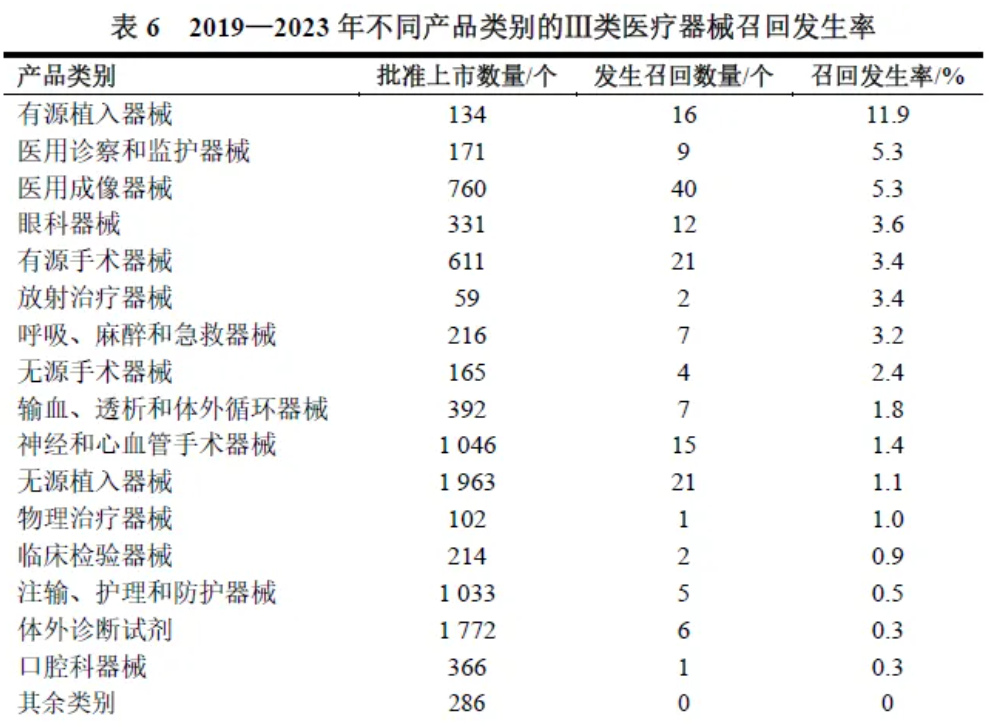
2019—2023年,国家药品监督管理局共批准上市9621个Ⅲ类医疗器械,其中169个曾经发生召回,Ⅲ类医疗器械的5年召回发生率为1.8%。
在169个召回的Ⅲ类医疗器械中,9.5%为一级召回,49.1%为二级召回,41.4%为三级召回。在Ⅲ类医疗器械中上市获批数量最多的是无源植入器械,其次是体外诊断试剂、神经和心血管手术器械,分别为1963个、1 772个和1046个。在Ⅲ类医疗器械中,最易发生召回的产品类别是有源植入器械,召回发生率达11.9%。与无源植入器械相比,有源植入器械的召回风险更高。见表6。
在2019—2023年获批的Ⅲ类医疗器械中,创新医疗器械共196个,Ⅲ类创新医疗器械的召回发生率《(1.0%)比非创新医疗器械的召回发生率《(1.8%)低。发生召回的2个创新医疗器械为进口Ⅲ类医疗器械,分别为标签问题的三级召回和软件原因的二级召回,发生召回时间差为1595d和304d。
发生一级召回的医疗器械特征
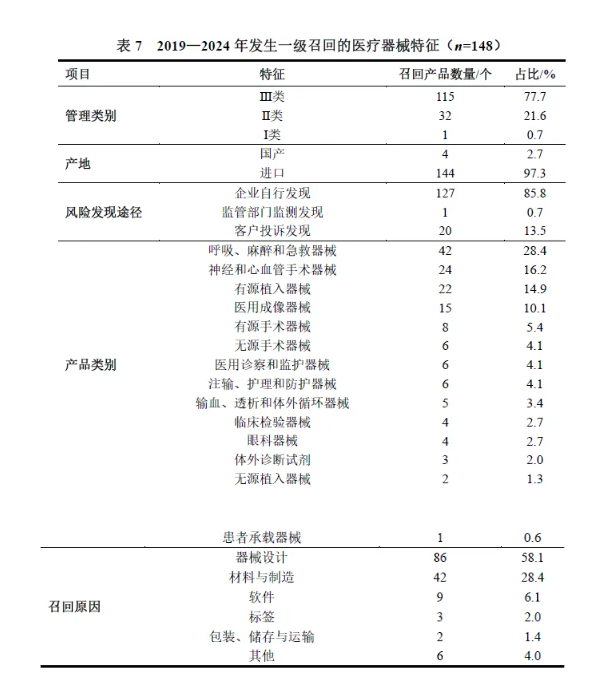
虽然一级召回仅占所有召回事件的6.0%,但其对患者安全的潜在影响最大,因此备受临床医生、生产商和监管机构的关注。2019—2024年,发生一级召回的医疗器械共148个,77.7%为Ⅲ类医疗器械,进口医疗器械占比远高于国产,85.8%为企业自行发现潜在风险后召回。发生一级召回的医疗器械产品类别主要为呼吸、麻醉和急救器械(28.4%)、神经和心血管手术器械(16.2%)和有源植入器械(14.9%)。医疗器械发生一级召回的主要原因是器械设计(58.1%)、材料与制造(28.4%)和软件(6.1%),见表7。
讨论
1-监管强化与企业责任意识共同推动我国医疗器械召回数量降低
我国医疗器械上市数量不断增加,创新程度不断提升,医疗器械召回数量逐年减少。2022年,我国成为全球医疗器械第二大市场,产品上市数量和创新程度明显增长,2019—2024年国内Ⅱ类、Ⅲ类医疗器械和创新医疗器械首次注册数量年复增长率为17.1%、17.7%和27.8%。但是,医疗器械召回数量开始逐年降低,我国医疗器械上市数量变化与召回数量变化未呈现相关性。说明我国监管力度逐渐加强,监督检查力度加大并且监督抽检精准高效。《医疗器械监督管理条例》等法规的持续修订和细化,对医疗器械的生产、销售和使用行为进行了更严格的规范。中国药品监督管理研究会发布的《中国医疗器械行业发展报告《(2024)》
显示,2023年省市区药品监督管理局组织飞行检查医疗器械生产企业4056家次同比增加6.32%,监督抽检不合格率同比下降0.9%。此外,医疗器械召回数量的减少还可能与企业质量意识提升和技术创新投入增加有关。
与进口医疗器械相比,国产医疗器械召回数量偏少。我国上市的医疗器械数量是进口的15.5倍,但是医疗器械召回的产品中有75.1%是进口医疗器械。这可能是因为进口医疗器械多为大型的跨国企业,为维护品牌形象与市场地位,对医疗器械产品执行高标准、严要求,发现潜在风险便积极主动召回。我国医疗器械生产企业中小型企业占比大、行业集中度低,企业的产品主体责任意识较为薄弱。
2019—2023年,Ⅲ类医疗器械的5年召回发生率为1.8%。医疗器械的召回发生率高或低不能简单理解为医疗器械安全或不安全。医疗器械召回发生率反映制造商遵守监管机构制定的监管标准的情况,较低的召回发生率可能表明医疗器械更安全、质量更高。但也可能存在低估风险的情况,如果医疗器械生产企业主动发现风险的能力较弱,即使发现风险也可能存在召回顾虑,企业不积极召回将造成召回发生率偏低。
2-鼓励国内企业加强不良事件上报,提升召回主动性
目前我国医疗器械召回主要依赖监管监测来发现缺陷。在国产医疗器械召回原因中,有61.6%是监管部门监测发现不合格后启动召回,在进口医疗器械召回原因中,有91.4%为企业自行发现潜在风险后召回。
根据国家药品监督管理局对《医疗器械召回管理办法》的解读,主动召回是指医疗器械生产企业按照有关要求或根据产品不良事件等信息对生产的医疗器械产品进行质量评估,确定医疗器械产品存在缺陷的,由生产企业主动实施的召回,是企业的法定义务。同样,美国21CFR7.40认为召回是一种自愿行为,因为制造商和分销商履行其责任,保护公众健康和福利免受具有伤害风险、严重欺骗性或者存在其他缺陷的产品影响。制造商或分销商可随时自愿发起召回。
医疗器械召回的起因很大程度上来自于不良事件监测发现的风险。根据国家药品监督管理局药品评价中心《(国家药品不良反应监测中心)发布的《国家医疗器械不良事件监测年度报告(2023年)》数据显示,2019—2023年的不良事件报告数量逐年上升,在2023年国家药品监督管理局药品评价中心(国家药品不良反应监测中心)收到的86万余份医疗器械不良事件报告中,89.38%为使用单位上报,8.42%为经营企业上报,注册人上报仅占比2.16%。同比2022年,使用单位上报的不良事件占比增加,经营企业和注册人上报占比减少。
生产企业主体责任意识欠缺,对医疗器械不良事件风险管理重视程度不足,企业主动上报不良事件的积极性有待加强。应加强国内医疗器械生产企业与医疗机构、医护人员的联动,建立便捷的沟通反馈渠道。加大对生产企业的培训,引导企业走出认识误区,积极上报不良事件,提升召回主动性。
3-对于具有高风险的医疗器械应当加大监测力度
在2019—2024年召回的医疗器械中50.4%为高风险的Ⅲ类器械,对其需采取特别措施进行严格控制,以实现安全管理和质量管理,突出溯源管理。
Ⅲ类医疗器械召回特征分析结果显示,召回发生率排名前3的产品类别是有源植入器械、医用诊察和监护器械、医用成像器械。应针对召回发生率较高的产品类别,加大监测力度和风险分析,开展专门的培训,采取更有效的管控措施以减少不良事件发生。
4-基于历史召回数据分析的风险预防体系可优化医疗器械设计与监管
国内医疗器械一级召回的主要原因分别是器械设计(70.1%)、材料与制造(14.9%)和软件《(8.8%)。
一项对美国食品药品监督管理局2018—2022年医疗器械一级召回的最常见原因的研究结果显示,召回原因主要是器械设计问题(54.5%),其次是制造错误《(13.2%),加工错误《(11.6%)和软件错误《(10.6%)。
器械的设计、材料选择以及制造过程中的缺陷,历来是导致医疗器械召回的关键因素。这些缺陷可能源于测试不充分、生物相容性考量不足,或是在设计过程中未能充分考虑人为因素及可用性,这些问题均有可能最终引致设计失败,对终端用户的安全性和制造商的声誉造成负面影响。
随着科技的发展,软件已成为大多数医疗器械不可或缺的组成部分。因此,编程异常、安全漏洞和网络连接问题等软件故障构成的软件设计问题成为医疗器械召回的主要原因,导致医疗器械召回数量不断增加[24,25]。通过分析已经发生召回的同类产品不良事件,可在器械设计阶段将风险因素纳入,以避免该类事件再次发生。
例如,植入类医疗器械与生物相容性《(金属毒性)是多例召回事件的重要原因,在设计研发同类产品时,应着重考虑选材的金属或其他材质特性、耐磨度等因素。应建立综合风
险数据库,通过大数据分析已发生的医疗器械召回事件,识别常见设计缺陷并制定预防措施。在产品设计初期引入《设计失效模式与影响分析《(design failure mode and effects analysis,DFMEA)”,促进跨部门合作,确保潜在风险得到全面评估和有效管理。要求企业设计新产品时务必依循综合风险数据库展开风险评估,细致比对同类产品曾经出现的风险因素,拟定风险防控设计方案。
5-医疗器械召回的企业主体责任尚需强化
《医疗器械监督管理条例》第六十七条规定,医疗器械注册人、备案人发现生产的医疗器械不符合强制性标准、产品技术要求,或者存在其他缺陷的,应当立即停止生产,召回已经上市销售的医疗器械,并将医疗器械召回和处理情况向负责药品监督管理的部门和卫生主管部门报告。《医疗器械召回管理办法》也同样明确企业是控制与消除产品缺陷的责任主体,应当主动对缺陷产品实施召回。
医疗器械生产企业应按照规定建立健全医疗器械召回管理制度,收集医疗器械安全相关信息,对可能的缺陷产品进行调查、评估,及时召回缺陷产品。此外,医疗器械生产企业应按照规定建立健全医疗器械质量管理体系和医疗器械不良事件监测系统。但实际上,目前国内企业的医疗器械召回管理制度、医疗器械质量管理体系和医疗器械不良事件监测系统还未充分发挥作用。
国内不良事件的报告主体来源主要是使用单位和经营企业,召回风险发现途径也主要是监管部门监测,因此国内医疗器械召回大多属于被动状态下的主动召回。国内普遍认为产品召回等同于产品质量低劣,或者将缺陷产品等同于非法产品。
国内企业对医疗器械召回存有顾虑和错误认知,认为召回会影响企业形象,波及企业股价以及收益。
国际上对缺陷产品的召回具有较高的接受度,认为医疗器械召回是企业负责任的行为,也是监管机构大力倡导的。
注,本文引自《2019—2024年我国医疗器械上市与召回风险分析及对策研究》原文链接https://link.cnki.net/urlid/3 1.2200.r.20250326.1228.002
本文2025-04-13 16:54:37发表“医休观点”栏目。
本文链接:https://www.yixiuqixie.com/article/735.html
相关文章









阅读排行
- 加速注射笔产能布局,英捷信医疗顺利完成数千万元A轮融资
- 康复住院病案首页现存问题:主要诊断选择错误、遗漏、不规范、手术操作编码遗漏、入院病情与出院诊断不一致
- 健适医疗完成2500万美元战略融资,进一步增强企业产品研发能力和发展运营模式
- 靠大单品融资超13亿元,这家关节植入物制造商16年只做一个产品
- 拥有28家康复医院的明州医疗,面对医保违规问题也束手无策
- 基于运动想象范式,搭建独特算法,韶脑科技有望将脑卒中康复周期缩短1/3
- 射频消融+左心耳结扎术后患者死亡,医院被索赔27.5万元
- 收藏!RDN一般手术操作步骤
- 房颤消融:“冷-热-光-电”的交相辉映
- 三明试点某康复科工分表曝光,一小时绩效5块钱!网友辣评:我还不如去干东郊到家