


魅丽纬叶RDN导管审评报告

本文转自公众号:医休RDN、医休器械(yixiuqixie.com)
大家好,我是医休哥,
技术审评概述
一、产品概述
(一)产品结构及组成产品由显影头中心拉丝、连接件、网篮支架、消融电极、网篮保护鞘、七腔管、手柄、接插件组成。
(二)产品适用范围产品在医疗机构使用,与本公司生产的肾动脉射频消融仪(型号:GL-06E15WA,软件发布版本:RDN-V1)配合使用,适用于辅助治疗难治性高血压及药物不耐受的高血压患者。其中难治性高血压定义为服用3种以上降压药物(含一种利尿剂)治疗3个月以上血压控制不佳的患者;药物不耐受是指药物禁忌、或因药物不良反应而不能耐受服药的患者。
(三)型号/规格GL-6W 8mmGL-6W 12mm
(四)工作原理本产品为用于经皮介入方式进行肾动脉消融的消融导管,产品前端为网篮状设计,可通过手柄操控扩张和收缩;含有共计6个电极用于传递射频能量,每个电极上均有测温传感器,可用于测量组织温度。
本次申报2个型号,区别在—5—于可适配的血管直径不同。当与本公司生产的肾动脉射频消融仪配套使用时,可将射频能量经过电极传递至肾动脉血管内膜,利用电流热效应使得肾动脉血管及外层神经热凝坏死,从而阻断交感神经的兴奋传导,来实现降低患者血压的目的。
二、临床前研究概述
(一)产品性能研究
申请人提供了产品性能研究资料以及产品技术要求的研究和编制说明,给出了外观、尺寸、物理性能(峰值拉力、弯曲疲劳、射线可探测性)、电学性能、化学性能、无菌、内毒素、微粒污染、网篮特性、电气安全和电磁兼容等功能性、安全性指标的确定依据。产品技术要求中各指标参考了相关的行业标准,包括:YY 0778-2018、YY 0285.1-2017。
(二)生物相容性
申请人依据GB/T 16886.1-2011对成品中与患者直接接触的导管和电极的生物相容性进行了评价。所评价材料短时接触人体循环血路,实施了生物学试验(细胞毒、致敏、皮内反应、急性毒性、热原、溶血、凝血、血栓形成),提交了医疗器械检验机构出具的生物学试验报告。因产品采用两种不同的粘合剂,分别选择两种工艺的产品开展了相应的生物相容性试验。
(三)灭菌
—6—产品由生产企业委托第三方进行环氧乙烷灭菌,无菌保证水平为10-6,申请人依据ISO 11135:2014标准按照半周期方法进行灭菌确认,提交了灭菌确认报告。采用强制解析方式去除残留,提交了EO和ECH的残留量测试报告。
(四)产品有效期和包装
产品货架有效期3年,申请人采用加速老化方式进行有效期验证,共计开展了等效时长3.5年的加速老化,并对老化后产品进行包装、无菌和各项性能测试,结果均符合要求。
申请人还提供了包装和模拟运输的验证资料,包括跌落、堆码、振动等各项测试,产品包装结构无破损。
(五)动物研究
1.申请人提交了基于离体猪肝开展的量效关系研究资料。
分别选择不同的消融时间(60/90/120/150s)和消融温度(55/60/70/80/90℃)进行测试,观察不同输出条件下组织消融深度和消融面积,并基于相关临床文献中关于肾动脉周围交感神经分布深度的数据,最终确定了临床使用的推荐治疗参数(60℃、120s)。
2.申请人提交了基于活体犬开展了动物试验资料。
试验在第三方机构开展,选择构建高血压模型的拉布拉多犬进行试验,共计纳入18只动物分为试验组和空白对照组,选择3个时间点(7/30/60天)进行随访观察,试验组分别在左右肾动脉进行射频消融,对照组仅进行肾动脉造影假手术,分别—7—在基线和术后不同时间点观察动物血压变化情况,以及解剖后的神经纤维损伤程度和血管内膜等部位变化情况。
结果表明,试验组动物在不同时间点的收缩压和舒张压均有较大程度的下降,对照组动物仅小幅度下降或升高;试验期间动物均无异常表现,未观察到肾动脉狭窄和夹层等情况,肾动脉神经纤维在不同时间点均显现明显损伤且无明显差异。
(六)有源设备安全性指标
产品符合GB 9706.1-2020的相关通用要求和GB9706.202-2021的专用安全要求,符合YY 9706.102-2021的电磁兼容并列安全要求,提供了医疗器械检验机构出具的配合本公司肾动脉射频消融仪一同开展的检验报告。
(七)其他
申请人提交了产品治疗效果影响因素的分析资料。基于相关临床试验数据,分别对不同消融点数、消融点定位、贴靠程度、术者经验和学习曲线等可能影响治疗效果的各项因素进行整理分析,结果表明最终治疗效果血压下降数值均无明显差异。同时提供了临床治疗的标准化操作流程和相关注意事项。
三、临床评价概述
申请人选择临床试验路径进行临床评价,临床试验的设计为前瞻性、多中心、随机、平行对照、优效性设计,选择的对照组为假手术组,行肾动脉造影,重要的入选标准为
A—8—年龄在18周岁以上,65周岁以下;
B原发性高血压患者已服用2种或2种以上降血压药物至少4周,血压仍≥150/90mmHg且<180/110mmHg(舒张压和收缩压都达到),和24小时动态血压平均收缩压≥135mmHg等。重要的排除标准为单侧或双侧肾动脉形状结构不适宜做消融手术的患者(肾动脉狭窄超过50%、肾动脉瘤、肾动脉畸形、肾动脉直径<4mm或可治疗段长度<20mm);有肾动脉介入治疗史或接受过肾脏去神经手术的患者;估算肾小球滤过率(eGFR)<45mL/min/1.73㎡的患者;继发性高血压患者;
E24小时动态血压提示平均收缩压小于135mmHg等。临床试验共入组205例受试者,试验组、对照组比例为2:1。
临床试验的主要评价指标为术后6个月诊室收缩压(SBP)相对基线(导入期末入组前)水平变化情况,术后6个月动态血压相较于基线水平的变化情况,术后6个月诊室血压达标率。
次要评价指标包括术后6个月舒张压相对基线水平变化情况;包括术后6个月24小时动态血压相对基线水平变化情况;术后6个月收缩压在目标范围(90mmHg≤SBP<140mmHg)内的病人百分比;术后6个月收缩压下降≥5mmHg的比例。
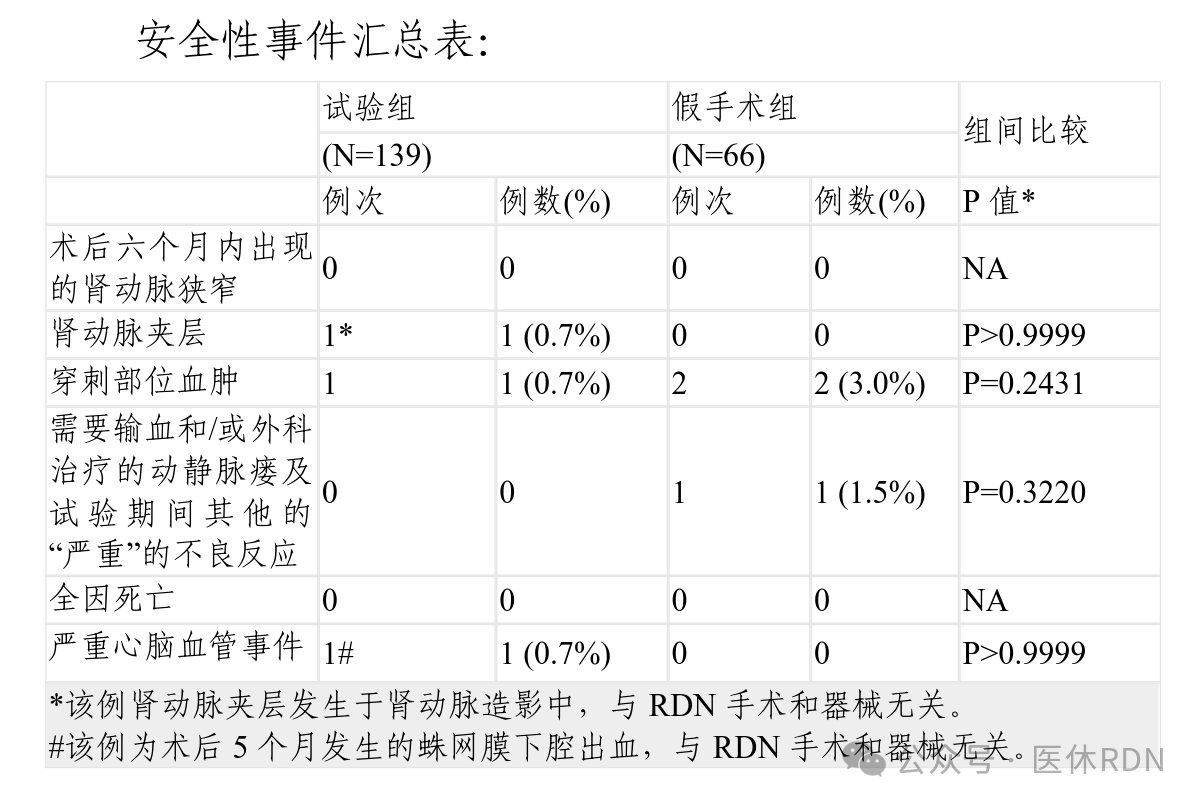
安全性评价指标为以参加试验的所有病例为对象,针对下述项目进行试验器械的安全性评价包括肾动脉狭窄、肾动脉夹层、脓血症、穿刺部位血肿、—9—术后六个月内出现新出现超过70%的肾动脉狭窄、需要输血和(或)外科治疗的动静脉瘘及试验期间其他的“严重”的不良反应、手术并发症、全因死亡、严重心脑血管事件、不良事件及严重不良事件等。
临床试验结果显示:
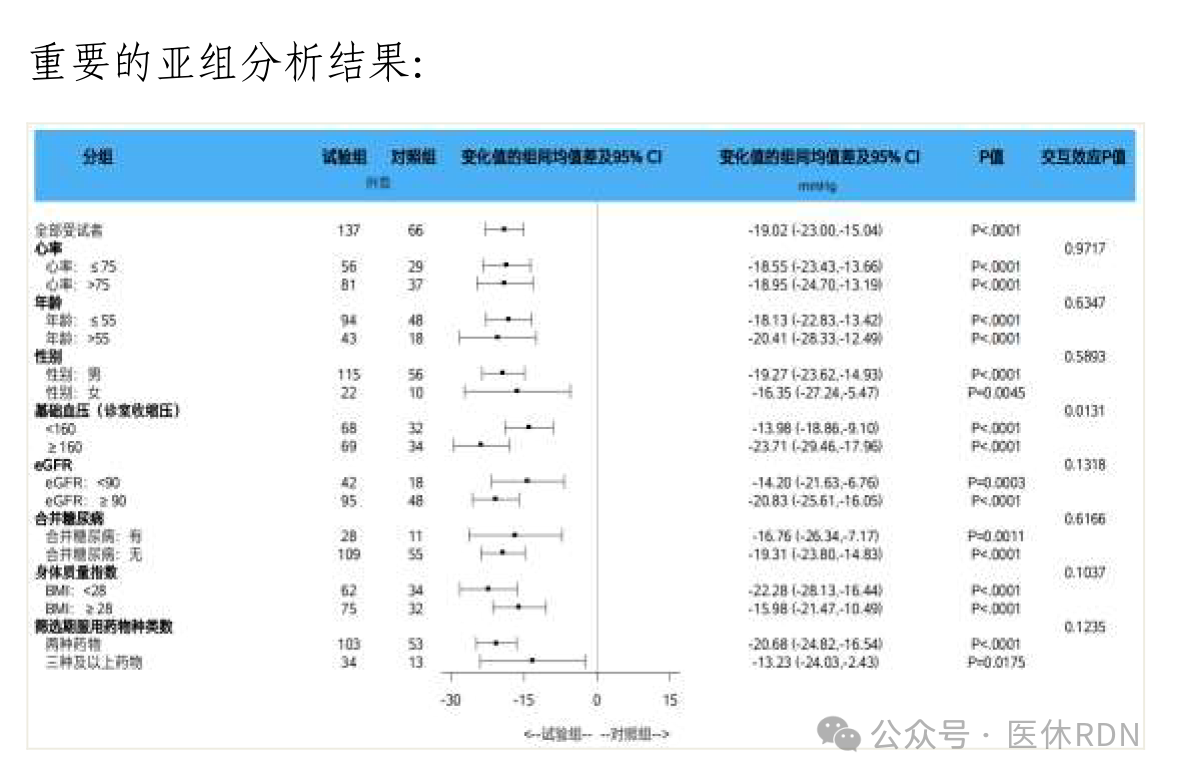
在FAS集,术后6个月诊室收缩压相对基线水平变化情况,试验组为25.2mmHg,对照组为6.2mmHg,组间差值为19.02mmHg,95%置信区间为[15.04mmHg,23.00mmHg],下限高于优效界值5mmHg;
在PPS集,试验组25.0mmHg,对照组6.2mmHg,组间差值18.79mmHg,95%置信区间为[14.73mmHg,22.85mmHg],下限高于优效界值5mmHg。
在FAS集,术后6个月动态血压相较于基线水平的变化情况,试验组12.51mmHg,对照组3.80mmHg,组间差值8.71mmHg,95%置信区间为[5.02mmHg 12.40mmHg],下限高于优效界值2.2mmHg;
在PPS集,试验组12.45mmHg,对组组3.50mmHg,组间差值8.95mmHg,95%置信区间为[5.19mmHg,12.70mmHg],下限高于优效界值2.2mmHg。
在FAS集,术后6个月诊室血压达标率,试验组为64.7%,对照组为7.7%,组间差值为57.0%,95%置信区间为[46.70%,67.33%],下限高于优效界值10%。
PPS集试验组为65.4%,对照组为7.8%,组间差值为57.6%,95%置信区间为[47.08%,68.07%],下限高于优效界值10%。
次要评价指标包括术后6个月舒张压相对基线—10—水平变化情况、术后6个月24小时动态血压相对基线水平变化情况、术后6个月收缩压在目标范围(90mmHg≤SBP<140mmHg)内的病人百分比、术后6个月收缩压下降≥5mmHg的比例等试验组均优于对照组,组间存在统计学差异。
安全性评价结果显示临床试验中未发生死亡或与RDN相关的肾动脉血管损伤(如肾动脉狭窄、夹层或穿孔),无因RDN器械或术后导致退出临床试验的情况发生,无研究器械相关的严重不良事件发生,在试验组和对照组间无显著差异。
 四、产品受益风险判定
四、产品受益风险判定
受益:
产品与本公司生产的肾动脉射频消融仪配合,适用于辅助治疗难治性高血压及药物不耐受的高血压患者。其中难治性高血压定义为服用3种以上降压药物(含一种利尿剂)治疗3个月以上血压控制不佳的患者;药物不耐受是指药物禁忌、或因药物不良反应而不能耐受服药的患者。
风险:
可能发生血管内神经消融已知不良事件,包括:血管并发症(例如,临床上显著的腹股沟血肿、动静脉瘘、假性动脉瘤、大出血),需要手术修复、介入手术、注射凝血酶或输血,需要介入的肾动脉穿孔,需要介入的肾动脉夹层,新发肾—12—动脉狭窄,严重栓塞事件,导致终末器官损伤等。
根据申请人提供的申报资料,经综合评价,在目前认知水平上,认为该产品的上市为适用人群带来的受益大于风险。考虑到上述产品受益和风险,对申报产品的适用人群进行了进一步的限定,用于辅助治疗难治性高血压及药物不耐受的高血压患者。
为保证用械安全,基于对主要剩余风险的规避,要求申请人在申报产品的说明书中提供了RDN治疗方法的患者选择说明及患者咨询的信息,需让患者充分了解产品的可能受益和风险,对患者进行充分告知,进一步控制产品临床风险。
本文2024-09-05 14:20:28发表“行业新闻”栏目。
本文链接:https://www.yixiuqixie.com/article/357.html
相关文章












阅读排行
- 手慢无!10款已上市密网支架·最全参数对比
- 湖北康复项目与医保支付目录
- 新源脑科学完成数千万元Pre-A轮融资,加速脑功能监测与神经调控产品落地
- 合珀生物完成数百万元天使轮融资,加速3D器官模型研发,助力革新药物研发进程
- 国家药监局关于暂停进口、经营和使用韩国杰希思医疗公司Nd:YAG激光治疗仪的公告
- 加速注射笔产能布局,英捷信医疗顺利完成数千万元A轮融资
- 健适医疗完成2500万美元战略融资,进一步增强企业产品研发能力和发展运营模式
- 靠大单品融资超13亿元,这家关节植入物制造商16年只做一个产品
- IVD新品:万孚、万泰、达安基因、东方生物、雅培等
- 基于运动想象范式,搭建独特算法,韶脑科技有望将脑卒中康复周期缩短1/3